The Renin-Angiotensin-Aldosterone System (RAAS) is a complex hormonal cascade that plays a pivotal role in regulating blood pressure, fluid balance, and electrolyte homeostasis in the human body. This article meticulously details the conversion of Angiotensin I to Angiotensin II and the subsequent downstream effects, illustrating how the kidneys, lungs, and adrenal glands collaborate to maintain cardiovascular stability. Understanding the RAAS is fundamental to comprehending the pathophysiology of hypertension and other cardiovascular and renal diseases.
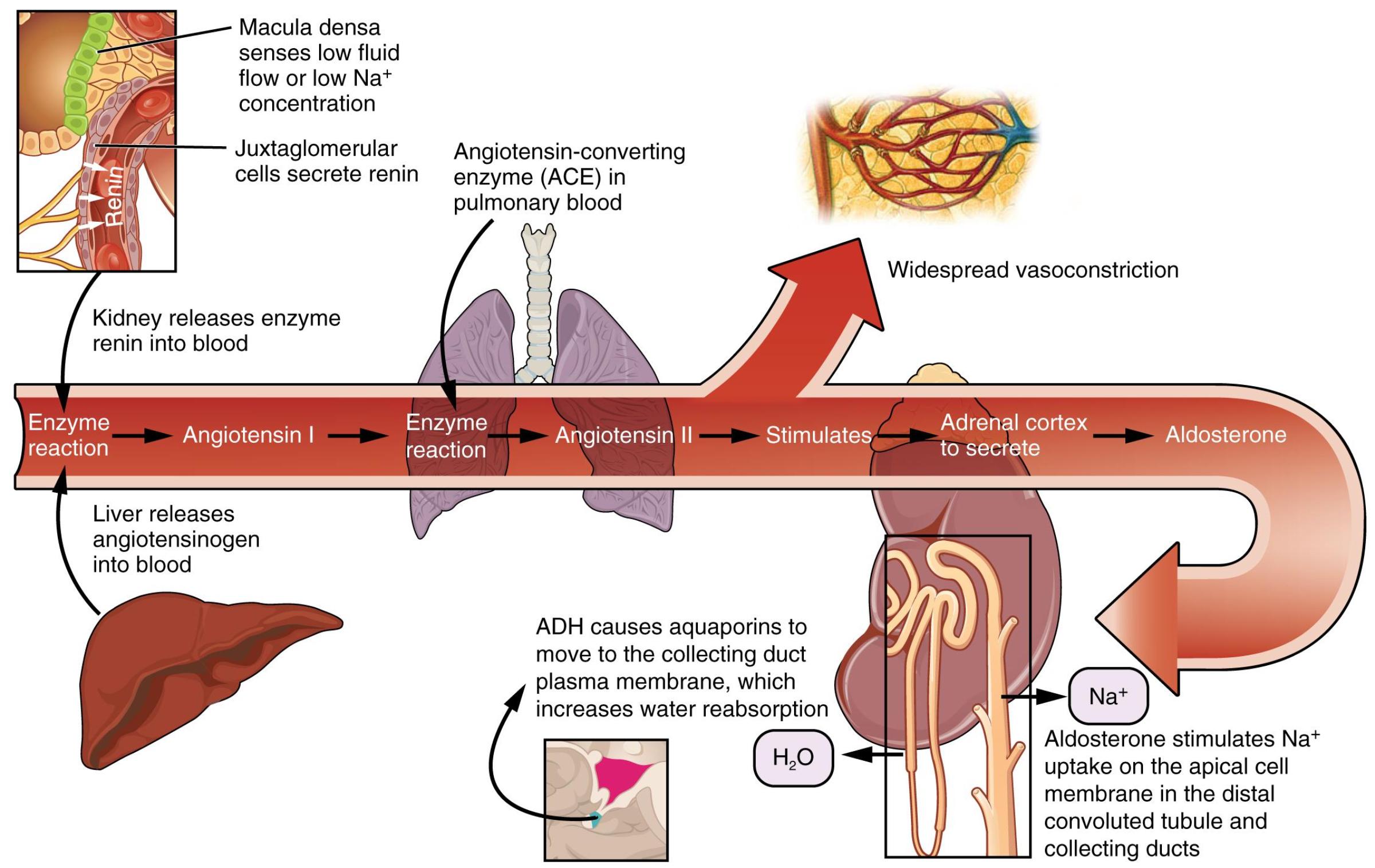
Macula densa senses low fluid flow or low Na+ concentration: These specialized cells in the distal convoluted tubule detect a decrease in the concentration of sodium ions or a reduced flow rate of tubular fluid. This signals the need for the body to retain more fluid and raise blood pressure.
Juxtaglomerular cells secrete renin: Located in the afferent arteriole, these cells respond to signals from the macula densa, decreased blood pressure, or sympathetic stimulation by releasing the enzyme renin into the bloodstream. Renin is the initiating factor of the RAAS.
Kidney releases enzyme renin into blood: The kidney, specifically the juxtaglomerular cells, acts as the primary organ for secreting renin. This crucial enzyme then circulates in the bloodstream to begin the cascade.
Liver releases angiotensinogen into blood: The liver continuously produces and releases angiotensinogen, a pro-hormone, into the bloodstream. Angiotensinogen is the precursor upon which renin acts.
Enzyme reaction (Angiotensin I): Renin, released by the kidney, cleaves angiotensinogen to produce angiotensin I. Angiotensin I is a relatively inactive decapeptide that serves as the substrate for the next step in the RAAS pathway.
Angiotensin-converting enzyme (ACE) in pulmonary blood: ACE is an enzyme found predominantly in the endothelial cells of the lungs, but also in other tissues. It is responsible for converting inactive angiotensin I into the highly potent active hormone, angiotensin II.
Enzyme reaction (Angiotensin II): ACE acts on angiotensin I to form angiotensin II. This conversion is a critical step, as angiotensin II is the main effector molecule of the RAAS, mediating most of its physiological actions.
Widespread vasoconstriction: Angiotensin II is a powerful vasoconstrictor, meaning it causes blood vessels throughout the body to narrow. This directly increases total peripheral resistance, leading to a rapid rise in blood pressure.
Stimulates Adrenal cortex to secrete Aldosterone: Angiotensin II also travels to the adrenal glands, where it stimulates the adrenal cortex to release aldosterone. Aldosterone is a mineralocorticoid hormone with significant effects on electrolyte balance.
Aldosterone: This hormone acts primarily on the kidneys. It plays a key role in regulating sodium and potassium levels, as well as maintaining blood volume and pressure.
Aldosterone stimulates Na+ uptake on the apical cell membrane in the distal convoluted tubule and collecting ducts: Aldosterone increases the reabsorption of sodium ions from the filtrate back into the blood in the DCT and collecting ducts. This process is often coupled with potassium secretion.
ADH causes aquaporins to move to the collecting duct plasma membrane, which increases water reabsorption: Antidiuretic hormone (ADH), also known as vasopressin, released from the posterior pituitary, acts on the collecting ducts. It promotes the insertion of aquaporin water channels into the membrane, leading to increased water reabsorption back into the bloodstream, thus concentrating the urine.
The Renin-Angiotensin-Aldosterone System (RAAS) is an elaborate hormonal system, fundamental to the long-term regulation of arterial blood pressure and extracellular fluid volume. It is activated primarily in response to decreased blood volume, reduced blood pressure, or sympathetic nervous system stimulation, signaling the body to conserve fluid and constrict blood vessels. This powerful regulatory cascade involves a series of enzymatic reactions and hormonal releases, demonstrating a remarkable example of inter-organ communication and physiological feedback. The diagram meticulously illustrates the sequence of events, starting with the kidney’s crucial role in initiating this cascade.
The RAAS begins when the juxtaglomerular apparatus in the kidney, particularly the macula densa and juxtaglomerular cells, detects a fall in blood pressure or a decrease in sodium delivery to the distal tubule. In response, the juxtaglomerular cells secrete the enzyme renin into the bloodstream. Renin then acts upon angiotensinogen, a protein produced by the liver and constantly circulating in the blood, converting it into angiotensin I. While angiotensin I has some weak biological activity, it is primarily a precursor to the main effector hormone. This initial step, catalyzed by renin, is the rate-limiting step of the entire cascade, making it a prime target for therapeutic intervention in hypertension.
As angiotensin I circulates in the blood, it encounters the angiotensin-converting enzyme (ACE), predominantly found on the surface of endothelial cells, particularly in the lungs. ACE swiftly converts angiotensin I into the highly potent octapeptide, angiotensin II. Angiotensin II is the active form of the hormone, exerting numerous effects that collectively work to raise blood pressure and conserve fluid. Its primary actions include causing widespread vasoconstriction, which directly increases total peripheral resistance, and stimulating the adrenal cortex to secrete aldosterone. Aldosterone, in turn, acts on the renal tubules, promoting sodium reabsorption and potassium excretion, which leads to increased water reabsorption and restoration of blood volume. Angiotensin II also stimulates the release of antidiuretic hormone (ADH) from the posterior pituitary, further enhancing water reabsorption in the collecting ducts, contributing to concentrated urine.
Dysregulation of the Renin-Angiotensin-Aldosterone System is a major contributor to several widespread medical conditions, most notably hypertension. Overactivity of the RAAS can lead to persistently high blood pressure, increasing the risk of cardiovascular events such as heart attack, stroke, and kidney disease. Pharmacological interventions targeting the RAAS are therefore cornerstones in the treatment of hypertension and heart failure. These include ACE inhibitors, which block the conversion of angiotensin I to angiotensin II, and angiotensin receptor blockers (ARBs), which prevent angiotensin II from binding to its receptors. By carefully modulating this complex system, medical professionals can effectively manage blood pressure and protect vital organs, highlighting the critical importance of understanding the RAAS in clinical practice and disease management.


{kind=link}